
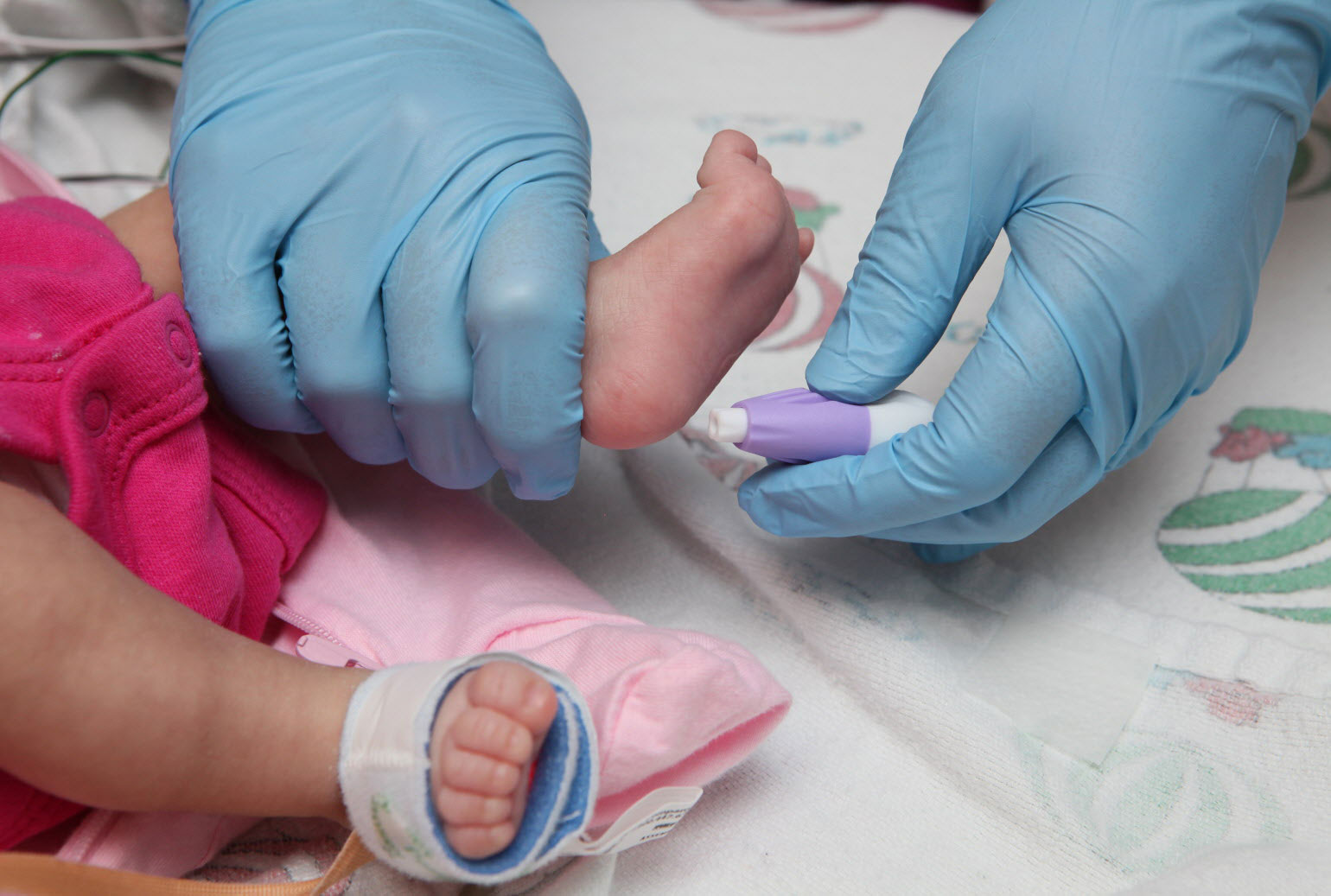
Dal 1992 tutti i neonati sul territorio nazionale italiano vengono sottoposti allo screening neonatale per l’individuazione di alcuni disordini metabolici, inclusa la fenilchetonuria o PKU.
Durante i primi giorni di vita, tra la 42esima e la 72esima ora dalla nascita, si raccolgono alcune gocce di sangue dal tallone del bambino tramite un piccolo prelievo. L’analisi del campione prelevato rivela la quantità di fenilalanina, un amminoacido contenuto nelle proteine, presente nel sangue del piccolo.
I test di screening per la fenilchetonuria alla nascita includono:
-
Test di Guthrie (BIA):
si tratta di un test economico, semplice e di facile utilizzo che viene applicato di routine. Una goccia di sangue viene posta su un disco di carta da filtro, detto spot, che viene fatto essiccare ed inviato al Centro regionale di screening di riferimento. Successivamente, viene messo a contatto con una coltura del batterio Bacillus subtilisa cui è stata aggiunta una sostanza, la β-2-tienialanina, che limita la crescita batterica. Se i batteri continuano a crescere, significa che sono presenti alti livelli di fenilalanina; di conseguenza è necessario che il neonato sia sottoposto a ulteriori analisi.
-
Analisi fluorometrica:
il sangue viene testato direttamente per alti livelli di fenilalanina tramite alcuni macchinari. I risultati sono più accurati rispetto a quelli ottenuti con la BIA.
-
Spettrometria di massa tandem (MS):
questo test ha un’affidabilità simile all’analisi fluorometrica e possono essere individuati anche i livelli di tirosina. La fenilalanina, infatti, viene scomposta in tirosina con l’aiuto dell’enzima fenilalanina idrossilasi. Questo enzima è carente nelle persone affette da PKU perciò questo test fornisce un quadro più completo e accurato della situazione.
-
Cromatografo per amminoacidi:
con questo test, accurato, si hanno meno “falsi positivi”. E’ un accertamento completamente gratuito e fa parte delle normali cure neonatali.
In generale, affinché i risultati dei test siano accurati, il neonato deve aver già avuto modo di assimilare alcune proteine come quelle contenute nel latte materno. Qualora il neonato fosse prematuro o sottopeso, infatti, i risultati potrebbero essere falsati: se il bambino è prematuro, il risultato potrebbe essere quello che viene definito “falso positivo” perché l’enzima per degradare la fenilalanina non si è ancora sviluppato completamente. Viceversa, potrebbe risultare un “falso negativo” se, allattato al seno o nutrito artificialmente, non stesse mangiando abbastanza, stesse vomitando o se il test fosse stato eseguito prima della 42esima ora dalla nascita. In questi casi l’esame potrà essere ripetuto.
Il risultato del test di screening non rappresenta una diagnosi definitiva.
Se il risultato è positivo indica il sospetto di PKU e in questo caso il centro di screening segnalerà il paziente al centro di cura il quale, a sua volta, provvederà ad effettuare ulteriori analisi biochimiche e molecolari per confermare o meno il disturbo. Tutti i pazienti malati vengono avviati alle terapie più idonee affinché possano condurre uno stile di vita normale, riducendo così le ospedalizzazioni e la mortalità precoce.
L´importanza di effettuare lo screening neonatale per la fenilchetonuria è legato al fatto che solitamente per i primi mesi di vita i neonati non manifestano sintomi, che compaiono invece successivamente.
Cos’è la Fenilchetonuria?
La fenilchetonuria, o PKU, è una malattia genetica che impedisce all’organismo di smaltire una proteina presente in alcuni alimenti.
Le proteine sono costituite da unità base chiamate amminoacidi: le persone affette da PKU non sono in grado di degradare l’amminoacido fenilalanina. Di conseguenza, la fenilalanina non metabolizzata si accumula nel sangue e nei tessuti provocando uno sviluppo anomalo del cervello con conseguente ritardo mentale.
La fenilchetonuria, se non curata fin dalla prima infanzia, causa disabilità intellettiva progressiva. Grazie alle terapie dietetiche adeguate e a un corretto controllo oggi i bambini affetti da PKU possono crescere e svilupparsi normalmente.
La fenilchetonuria è un disordine genetico
La PKU è una malattia metabolica ereditaria che si verifica quando un bambino riceve da entrambi i genitori due copie difettose del gene PAH. Il gene dice alla cellula di creare un enzima, la fenilalanina idrossidasi, che scomponga l’amminoacido fenilalanina. Se i geni sono difettosi e mal funzionanti (se sono quindi presenti delle mutazioni) inviano un messaggio scorretto. Nella fenilchetonuria, le cellule non producono l’enzima che scompone la fenilalanina, che pertanto si accumula nel sangue e nei tessuti.
La PKU è causata da un ampio numero di mutazioni nel gene PAH (12q22-q24.2); in letteratura sono inoltre descritte mutazioni di altri geni, che causano una condizione nota come iperfenilalaninemia da deficit di BH4.
In ogni cellula ci sono due copie per ogni gene: una copia che riceviamo da nostra madre e l’altra copia che riceviamo da nostro padre. Chi possiede una copia normale e una copia difettosa del gene PAH, è portatore della malattia. I portatori di PKU sono sani perché la copia normale sovrascrive il gene difettoso. Ciò significa che le cellule producono abbastanza enzimi per prevenire l’accumulo di fenilalanina.
Quando entrambi i genitori sono portatori del gene PAH difettoso, il loro bambino nascerà con PKU poiché riceve una copia del gene difettoso da ciascun genitore. Quando entrambi i genitori sono portatori, le possibilità ad ogni gravidanza sono:
- 1 su 4 di avere un figlio malato di PKU
- 2 su 4 di avere un bambino che è un “portatore”
- 1 su 4 di avere un bambino sano.
Fenilchetonuria e dieta
Il modo principale per trattare la PKU consiste nel seguire una dieta speciale che limiti gli alimenti contenenti fenilalanina. Nel neonato vengono somministrati idrolizzati o miscele di amminoacidi puri. Durante la crescita, è necessario evitare cibi ricchi di proteine come: uova, formaggio, noccioline, latte, fagioli, pollo, manzo, maiale, pesce.
Per assicurarsi che ricevano una quantità adeguata di proteine, è necessario che le persone affette da PKU seguano una dieta rigida per tutta la vita. Per questo motivo, le persone affette da PKU devono lavorare a stretto contatto con un medico o un dietologo per mantenere un corretto equilibrio di nutrienti e limitare l’assunzione di fenilalanina. Devono anche monitorare i livelli di fenilalanina assunti durante il giorno tenendo dei registri alimentari.
Il livello di mantenimento raccomandato è di solito tra i 120 e i 360 µmol/L nei neonati, mentre nei pazienti più grandi i valori superano i 600 µmol/L. Non esiste, però, un livello soglia di riferimento della fenilalanina oltre al quale si deve cominciare il trattamento.
Le persone affette da PKU sottoposte ad una dieta a basso contenuto di fenilalanina e amminoacidi misti e ad una terapia farmacologica possono crescere e svilupparsi normalmente.
Trattamento farmacologico per la fenilchetonuria
Nel 2015 la Food and Drug Administration (FDA) ha approvato la sapropterina (Kuvan) per il trattamento della PKU. La sapropterina aiuta a ridurre i livelli di fenilalanina. Questo farmaco deve essere usato in combinazione con un piano alimentare specifico. Tuttavia, non funziona allo stesso modo per tutti i malati, è più efficace nei bambini affetti da fenilchetonuria lieve.
Attualmente, BioMarin Pharmaceutical ha ottenuto l’autorizzazione all’immissione in commercio negli USA di un prodotto (pegvaliase) per il trattamento degli adulti affetti da fenilchetonuria. L’EMA (Agenzia Europea per i Medicinali) ha accettato all’inizio del 2018 di esaminare la richiesta della casa farmaceutica per ottenere l’approvazione della terapia.
Quali sono i centri di cura per la fenilchetonuria?
L’elenco è disponibile a questo link.
Per saperne di più visita anche la sezione FENILCHETONURIA di Osservatorio Malattie Rare.
Notizie recenti sullo screening per la fenilchetonuria
- Galattosemia Classica: nel Regno Unito identificata incidentalmente allo screening per la fenilchetonuria
 In base a uno studio retrospettivo, il 39% di diagnosi di GC è stata fatta indirettamente attraverso NBS Nel Regno Unito, la Galattosemia Classica (CG) viene identificata incidentalmente dallo Screening Neonatale (NBS) per la fenilchetonuria (PKU) utilizzando un percorso di “Sospetto di altro disturbo”
In base a uno studio retrospettivo, il 39% di diagnosi di GC è stata fatta indirettamente attraverso NBS Nel Regno Unito, la Galattosemia Classica (CG) viene identificata incidentalmente dallo Screening Neonatale (NBS) per la fenilchetonuria (PKU) utilizzando un percorso di “Sospetto di altro disturbo” - Screening neonatale, l’Assemblea Nazionale francese approva l’uso dei test genetici
 Oggi in Francia i neonati vengono sottoposti a screening solo per cinque malattie, ma l’Autorità Nazionale per la Salute raccomanda di includerne altre otto Parigi (Francia) – La Francia ha finalmente promosso una norma che permetterà l’uso dei test genetici all’interno del proprio programma
Oggi in Francia i neonati vengono sottoposti a screening solo per cinque malattie, ma l’Autorità Nazionale per la Salute raccomanda di includerne altre otto Parigi (Francia) – La Francia ha finalmente promosso una norma che permetterà l’uso dei test genetici all’interno del proprio programma